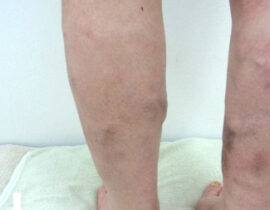
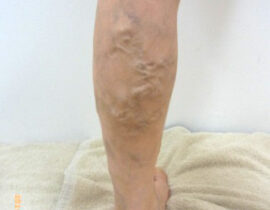
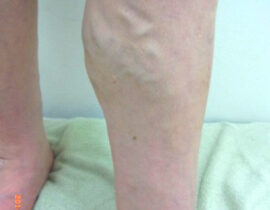
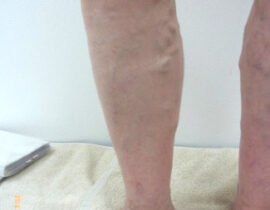
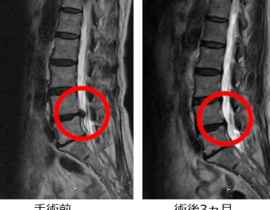
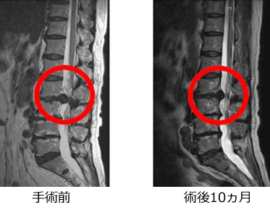
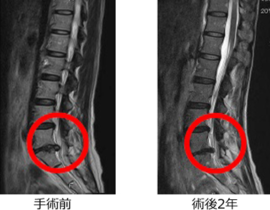
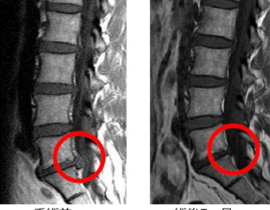
遺伝子治療概要
目次
1.CDC6shRNA治療(CDC6 RNAi)概要
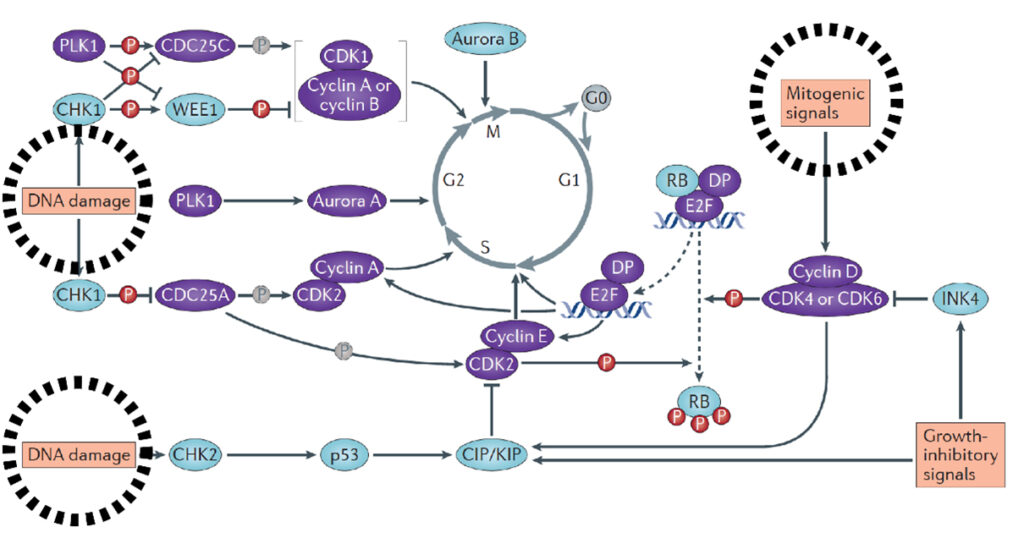
がん細胞の無秩序な増殖は細胞周期が正常に制御されなくなるために生じ、その主たる原因として、①細胞周期エンジンを構成する分子群(及びその遺伝子座)の変異、②細胞周期の制御に関与する細胞内プロセスの異常、などが挙げられます。故に細胞周期エンジン構成分子群及び細胞周期制御に関与する細胞内プロセスはがん治療の有望なターゲットとなります。
細胞周期はcyclinsやcyclin-dependent kinases(CDKs)などの細胞周期エンジンを構成する分子群により高度に調節されており、多くのがん細胞において、このような分子群が過剰に活性化されています。また特定の刺激によりがんが発症する発がん動物モデルにおいて、これらの構成分子の働きを薬理学的および遺伝学的に阻害すると、がんの発症・進展が有意に抑制されることも明らかとなっています。興味深いことに、がんの種類によって、どの構成分子を阻害すると制癌効果があるのかが異なっています。これらの知見は、異なる種類のがん細胞の増殖は特定の細胞周期エンジン構成分子にそれぞれ特異的に依存しており、それら構成分子の作用を選択的に阻害することにより、多種多様ながんの治療が可能になることを示唆します(Nat Rev Cancer. 2017;17(2):93-115)。
実臨床では、2015年に細胞周期エンジン構成分子を標的とした抗がん剤として世界で初めて、CDK4/6選択的阻害剤であるpalbociclib(IBRANCE®: Pfizer)がER+/HER2-の閉経後進行乳がんを適応症として米国FDAによる条件付き承認されました。また現在、CDK4/6選択的阻害剤(乳癌、肺癌)、Polo-likeキナーゼ阻害剤(骨髄異形成症候群、膵癌)、Aurora阻害剤(T細胞リンパ腫)などの細胞周期エンジン構成分子を標的としたがん治療薬が臨床試験第3相まで進んでいます。
このように、細胞周期の制御はがん治療における有効なストラテジーの一つとして既に確立されており、細胞周期エンジンを構成する分子群を対象にした分子標的療法に関しては研究が進んでいます。一方、細胞周期に影響を及ぼす細胞内プロセスを標的とした治療法については研究が不十分なため、同治療法は革新的ながん治療に繋がる余地が残されています。そのような細胞内プロセスとして重要な「DNA複製ストレス(replication stress)及びそれにより惹起されるDNA損傷反応(DNA damage response:DDR)」にフォーカスした「次世代がん治療」としてCDC6 RNAi はこれからの期待される治療法の一つと考えられます(図1)。
2.CDC6 RNAi の作用
がんの特徴の最たるものが、無秩序に無限に増殖する=無限に細胞分裂することです。細胞が分裂する際には、細胞核内の染色体の倍加すなわちDNAの複製が必要です。その際に、licensing factor(ライセンシングファクター)と呼ばれるタンパクやタンパク複合体が必須なのですが、がん細胞にはその種類に関わらずこのライセンシングファクターが無秩序に大量に発生していることが確認されており、それが、がんの無限増殖を促しています。
ライセンシングファクターの最たるものであるCDC6タンパクが、前がん細胞やがん細胞において過剰発現しているため、本来はライセンシングファクターが制御する細胞内プロセスが破綻しています。その過剰発現しているCDC6タンパクを減少もしくは消去できれば、がん細胞の分裂=増殖が止まり、ひいては、がん細胞の自然消失(自殺)が期待できます。
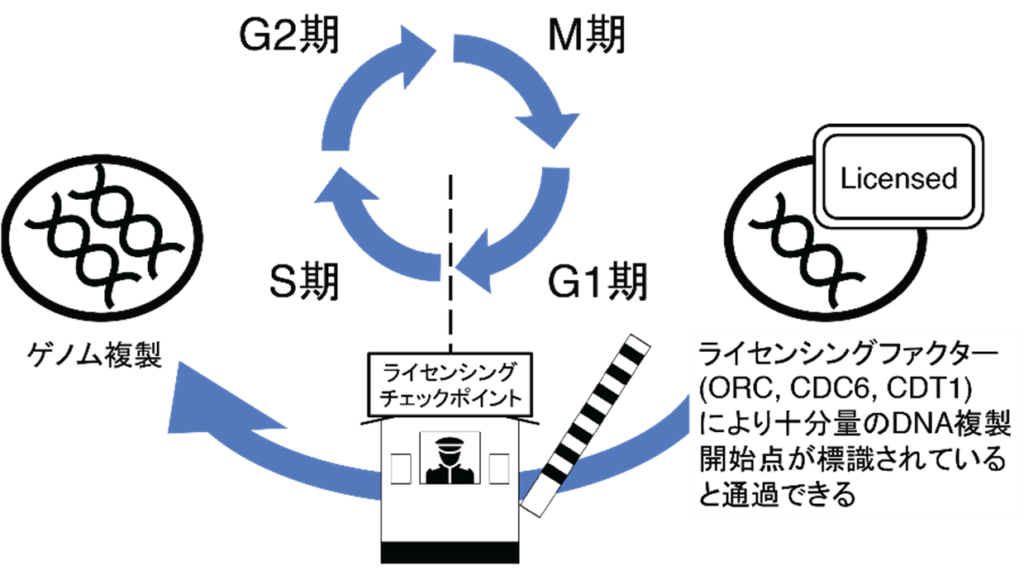
遺伝子治療のひとつCDC6 RNAi(CDC6 RNA干渉)は、このCDC6タンパクの合成に必要ながん細胞核内のメッセンジャーRNAを消去してCDC6タンパクの合成を阻害し、それによりCDC6タンパクの消失を目指すものです。がん細胞の分裂に必須なCDC6を消去して、がんの分裂増殖を止め、結果としてがんの自殺誘導を導くことを意図しています(図2)。
それを実現させるためには、前がん細胞やがん細胞内に大量に発生しているCDC6タンパクを生み出している核内のメッセンジャーRNAに働きかける特殊なマイクロRNAを細胞核内に届けなければいけません。その運搬体として、病原性を除去したウイルスベクターを用い、それに治療に必要な遺伝子(マイクロRNA)を搭載して体内に投与します。本遺伝子治療で採用しているベクターはレンチウイルスベクター※でがん細胞以外の細胞にも侵入する場合がありますが、がん細胞に侵入した時のみ遺伝子発現が生じる仕組みが施されており正常細胞には何ら問題となる反応が生じない設計になっています。
このベクターには、CDC6タンパクを消去するためのマイクロRNA(CDC6shRNA)の他に、多くのがん細胞で欠失しているがん抑制遺伝子群であるP16、PTEN、P53も搭載してがん細胞の分裂抑止効果を高める工夫を施しています。
◆図2:G1期とS機の間に存在するライセンシングチェックポイント
3.非増殖型レンチウイルスベクターの不活化型ウイルス相当性
非増殖型レンチウイルスベクター(Replication-Incompetent Lentiviral Vector, RILV)は、自己増殖能力を欠くように設計されており、以下の点で不活化ウイルスに相当すると解釈できる。
- 自己複製能力の欠如 通常のレンチウイルスは宿主細胞に感染し、自身の遺伝情報を細胞のゲノムに組み込んで複製する。しかし、非増殖型レンチウイルスベクターは以下のような改変が施され、自己複製ができないようになっている。
- 生物学的・環境リスクの低減 環境中での持続性の欠如: 自己複製しないため、一度細胞に感染しても、他の細胞へ感染拡散することはない。
- 不活化型ウイルスとの類似点 不活化型ウイルスとは、物理的・化学的処理(熱処理やホルムアルデヒド処理など)により感染能力や複製能力を失わせたウイルスを指す。一方、非増殖型レンチウイルスベクターも遺伝子工学的改変により複製能力を失っており、その意味で 「機能的に不活化された」ウイルス であると考えられる。
- 当核酸化合物の不活化処理について ― 製造元からの報告 ICH Q5A(R2)ガイドラインにおけるウイルス除去/不活化プロセスについて
・ウイルスの主要な複製遺伝子(gag, pol, env)が欠損しており、これらの遺伝子は製造時に一時的に供給されるが、ウイルス自体には残らない。
・パッケージング信号(Ψ)の除去または変異導入により、ウイルスゲノムが新たなウイルス粒子へ組み込まれない。
これにより、ウイルスは感染可能ではあるものの、宿主細胞内で新たなウイルス粒子を産生する能力を失っている。
ゲノムへの統合後の安定性: ウイルスゲノムが宿主細胞のゲノムに組み込まれた後は、宿主細胞内でのみ発現し、新たなウイルス粒子を作れない。
生物多様性への影響がないことを示すには「環境中で増殖・拡散するかどうか」 が基準となるが、下記の点で「不活化型ウイルス」と同様に、問題がないと判断できる。
①非増殖型レンチウイルスベクターは自己増殖しないため、環境中で拡散しない
→ 生物多様性に影響を及ぼさない。
②ウイルスゲノムは宿主細胞内でのみ発現し、新たなウイルス粒子を産生できない
→ 他の細胞に感染しないため、感染拡大のリスクがない。
③ 不活化型ウイルスと同様に「機能的に不活化」されており安全。
当社の製品製造システムの観点から体系的に説明いたします。
ICH Q5A(R2)「ヒト又は動物細胞株由来バイオテクノロジー製品のウイルス安全性評価」は、医薬品規制調和国際会議(ICH)が発行したガイドラインです。主に従来のバイオテクノロジー製品(組換えDNAタンパク質製品など)のウイルス安全性評価に焦点を当てており、サイトカイン、モノクローナル抗体(mAb)、サブユニットワクチンなど、組換えDNA技術を用いたin vitro細胞培養によって製造される製品も対象としています。
ICH Q5A(R2)ガイドラインでは、ウイルス汚染のリスクは細胞株由来のバイオテクノロジー製品全てが留意すべき事項であり、リスク源には以下の2つの側面があると強調しています。
(1):内因性ウイルス:由来細胞株(細胞マトリックス)自体からの汚染。生産細胞株の調製に用いられる動物が外因性ウイルスに感染しているか、またはウイルスを保有している場合、細胞株調製工程中に予期せぬウイルスの混入につながる可能性があります。
(2) 外因性ウイルス:生産工程中に偶発的に混入した外因性ウイルス、汚染された生物学的試薬または細胞培養原料(動物血清またはヒト血清など)の使用、生産環境が関連する規制要件を満たしていないことなど。
ICH Q5A(R2)ガイドラインでは、リスク要因に基づいて生産工程における外因性ウイルスおよび内因性ウイルスの除去能力を評価する必要があると規定されています。製品への内因性および外因性ウイルスの侵入リスクを確実に排除するため、主に以下の方法を採用しています。
1. 内因性ウイルスの侵入を防ぐため、以下の対策を講じています。
当社は米国サーモフィッシャー社と特許ライセンス契約を締結し、2018年に米国サーモフィッシャー社の生産細胞293Fを導入しました。その安全性の利点は、業界および当局から広く認められています。
① 293F細胞はSV40ラージT抗原を発現しないため、この抗原によって引き起こされる潜在的な腫瘍形成および免疫原性リスクを回避し、ウイルスパッケージング細胞に対する規制当局(FDAおよびCDE)の安全性要件を満たしています。
② 293F細胞株は、21 CFR 211およびEudraLex Volume 4に従って製造され、ICH Q5AおよびICH Q5Dに従って特性評価されています。複数のウイルス試験および継代試験を経て、細胞株には細胞バンクの設立、培養履歴、識別報告書などを含む完全なトレーサビリティ文書が付属しており、臨床レベルのアプリケーションの安全性を確保しています。
米国サーモフィッシャー社から生産細胞を導入することで、当社は生産システム全体において、生産源からの内因性ウイルス汚染が起こらないことを保証しています。
米国サーモフィッシャー社からの293F生産細胞の購入承認および出荷情報は次のとおりです。
| 製品番号: | A3152801 |
|---|---|
| 製品バッチ番号: | 556726-WCB1 |
| 輸送通関申請番号: | 260200002018051300 |
2. 外来ウイルスの侵入を防ぐため、以下の対策を講じています。
① 米国サーモフィッシャー社と特許ライセンス契約を締結しました。米国サーモフィッシャー社の製造セル293Fの導入に続き、同社の懸濁液システムの導入も継続しました。このシステムは無血清懸濁液製造システムであり、血清を導入しない製造方法を採用しているため、製造工程における外来ウイルスの混入を防止できます。また、米国サーモフィッシャー社から輸入した専用試薬を使用することで、ICHガイドラインの要件を満たし、低毒性、動物由来成分不使用、コンプライアンス、安全性といった大きな利点を有しています。
表は、米国サーモフィッシャー社と締結した特許ライセンス契約の内容を示しています。
ライセンス取得済み特許
| 出願番号: | 特許名: |
|---|---|
| 15/721,105 | レンチウイルス生産のための無血清懸濁液システム 米国 |
| PCT/US2017/54511 | レンチウイルス生産のための無血清懸濁液システム PCT |
| 62/402,877 | レンチウイルス生産のための無血清懸濁液システム 米国 |
② 研究開発プロセスの信頼性と安全性を確保するため、当社は800平方メートルの生産工場(GMP準拠レベル)を建設し、5つのC+Aグレードのクリーンエリア操作エリアを備えています。これらの工場には、WAVE25ウェーブバイオリアクター(Cytiva社、米国)、パルス式真空滅菌装置(Hualin Medical社)、生物学的安全キャビネット(Haier Biomedical社)、顕微鏡システム(OLYMPUS社、日本)、細胞カウンター(Thermo Fisher社、米国)、二酸化炭素インキュベーター(Thermo Fisher社、米国)、AKTA™ Pilot 600などを備えています。 (米国Cytiva社製)、AKTA™ flux 6 /flux S (米国Cytiva社製)などの生産設備に加え、すべての工場に独立したクリーン空調濾過システムを設置し、生産設備と生産環境がNMPA規制の要件を満たしていることを確保しています。
3. 生産プロセスへのウイルス除去/消火プロセスの追加
バイオ医薬品のウイルス除去プロセスは、製品の安全性を確保するための重要なプロセスです。各生産リンクを厳密に管理し、内因性および外因性ウイルスの侵入リスクを低減する条件下で、当社は製品の安全性をより確実に確保するために、製品生産プロセスの最終段階で低pH培養によるウイルス除去処理を追加しました。製品のpH値を4.0に調整し、緩衝液イオン濃度を調整し、4℃の低温培養プロセスを実施することで、脂質エンベロープウイルスのエンベロープタンパク質とカプシドタンパク質を破壊し、不活性化することで、ウイルス除去/不活化の要件を満たします。最終的に製品の安全性を確保します。
当社は、上記を通じて、米国サーモフィッシャー社の全生産システムを導入し、Cytivaや米国サーモフィッシャー社などの国際企業と技術設備提携を行い、先進的かつ安全な生産プロセスを確立しました。細胞株、生産設備、生産プロセス、生産原料および副資材の品質を厳格に管理し、製品が内因性および外因性のウイルス侵入リスクから解放されることを保証しています。